FRONTIERS IN MEDICAL CASE REPORTS - Volume 7; Issue 4, (Jul-Aug, 2026)
Pages: 01-09
Date of Publication: 07-Jul-2026
Print Article
Download XML Download PDF
Familial Characterization of the DSC2 c.354+1G>T Splice-Site Variant Cardiac Disease
Author: José Cedeño, Carlos Chellaram, Carolina Vega, Evelyn Medina, Omar A. Espinosa, Luis Méndez, Luis Sotillo, Lydier De Gracia
Category: Medical Case Reports
Abstract:
Arrhythmogenic cardiomyopathy is an inherited myocardial disease frequently associated with variants in desmosomal genes. DSC2 encodes desmocollin-2, a cadherin involved in cardiomyocyte adhesion at the intercalated disc; disruption of desmosomal integrity may contribute to myocardial remodeling and arrhythmogenic susceptibility. We describe a Panamanian family in which the DSC2 splice-site variant c.354+1G>T (NM_024422.6) was identified during evaluation for suspected inherited cardiac disease. The proband was a 66-year-old woman with hypertrophic cardiomyopathy, concentric hypertrophy on echocardiography, and high-risk syncope. Her 86-year-old mother had cardiac conduction disease requiring pacemaker implantation, and the proband’s daughter was identified as a carrier. The variant affects the canonical +1 donor splice site and is predicted to alter pre-mRNA splicing, with SpliceAI donor-loss score of 0.96, Pangolin splice-loss score of 0.64, CADD PHRED score of 33, and PhyloP score of 8.87. ClinVar classifies the variant as likely pathogenic. These findings support genetic counseling, cascade testing, and cardiovascular surveillance. However, the current clinical and segregation data do not establish definitive causality for hypertrophic cardiomyopathy or conduction disease.
Keywords: DSC2, Desmocollin-2, Desmosome, Splice-Site Variant, Inherited Cardiomyopathy, Hypertrophic Cardiomyopathy, Cardiac Conduction Disorder, Panama
Full Text:
Introduction
Arrhythmogenic cardiomyopathy is an inherited myocardial disease characterized by structural remodeling, ventricular arrhythmias, and an increased risk of sudden cardiac death. According to McNally, et al. (2023), the classic form, arrhythmogenic right ventricular cardiomyopathy, is associated with progressive myocardial replacement by fibrofatty tissue, predominantly affecting the right ventricle. However, Costa, et al. (2021) emphasized that left-dominant and biventricular forms are increasingly recognized, expanding the clinical spectrum of arrhythmogenic cardiomyopathy beyond the traditional right ventricular phenotype.
The molecular basis of arrhythmogenic cardiomyopathy is strongly linked to variants in genes encoding desmosomal proteins, including PKP2, DSP, DSG2, DSC2, and JUP. These proteins are essential components of the cardiac intercalated disc, where they contribute to mechanical adhesion between cardiomyocytes and to the maintenance of electrical stability during repeated myocardial contraction. DSC2 encodes desmocollin-2, a calcium-dependent cadherin located in the desmosome. Disruption of desmocollin-2 may impair cell-to-cell adhesion, alter desmosomal integrity, and contribute to myocardial remodeling, ventricular arrhythmias, and cardiomyopathic phenotypes.
Goudal, et al. (2022) reported that rare variants in arrhythmogenic cardiomyopathy genes contribute to the genetic burden of disease. However, interpretation of rare desmosomal variants remains complex, particularly when the clinical phenotype does not fulfill classical criteria for arrhythmogenic cardiomyopathy. Wu, et al. (2022) described deleterious rare desmosomal variants in patients with hypertrophic cardiomyopathy, suggesting that desmosomal genes may occasionally appear in overlapping or non-classical cardiomyopathy phenotypes. Similarly, Lin, et al. (2021) reported overlapping hypertrophic cardiomyopathy and left ventricular noncompaction associated with a DSC2 variant. These observations support the need for cautious genotype-phenotype interpretation, because the presence of a desmosomal variant does not automatically establish causality for hypertrophic remodeling or conduction disease.
A broader Panamanian cohort of hereditary cardiomyopathies previously reported pathogenic and likely pathogenic variants, including DSC2, in patients evaluated by targeted next-generation sequencing (Cedeño-Escudero et al., 2026). The present report focuses on the familial characterization of the DSC2 c.354+1G>T splice-site variant identified in a Panamanian family with heterogeneous inherited cardiac disease.
The aim of this article is to describe the clinical, familial, and molecular features associated with the DSC2 c.354+1G>T variant, while emphasizing the limitations of genotype-phenotype correlation and the need for expanded cardiovascular phenotyping and family segregation analysis.
Case Presentation and Family Study
Case Presentation
A Panamanian family with cardiovascular disease across multiple generations was evaluated in the context of suspected inherited heart disease. The proband was a 66-year-old woman with hypothyroidism, chronic pulmonary emphysema, and a clinical diagnosis of hypertrophic cardiomyopathy. She presented with high-risk syncope, and echocardiography showed concentric hypertrophy.
Family history documented that the proband’s mother, aged 86 years, had a cardiac conduction disorder requiring pacemaker implantation. Targeted family testing identified the DSC2 c.354+1G>T variant in the proband, her mother, and her daughter. The daughter was reported as a carrier during family evaluation. No definitive arrhythmogenic right ventricular cardiomyopathy phenotype was documented in the available data for the family members included in this report.
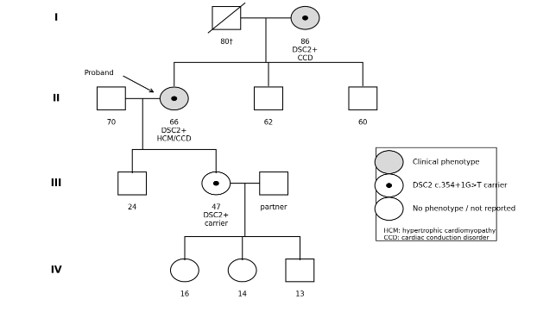
Figure 1: Four-generation pedigree of the family carrying the DSC2 c.354+1G>T variant. Shaded symbols indicate individuals with reported cardiovascular phenotype; central black dots indicate confirmed carriers of the DSC2 variant; open symbols indicate individuals without reported phenotype or not genetically tested. The arrow indicates the proband. HCM, hypertrophic cardiomyopathy; CCD, cardiac conduction disorder.
Sequencing, Bioinformatic Analysis, and Variant Interpretation
Genetic analysis in the proband was performed using next-generation sequencing with a capture-based cardiovascular gene panel designed for cardiomyopathies and inherited arrhythmias. The panel included 128 clinically relevant genes covering coding regions and splice-site boundaries, with an approximate target region of 470 kb. Library preparation was performed according to the validated laboratory protocol, and sequencing was carried out using a benchtop next-generation sequencing platform.
Sequencing reads were aligned to GRCh37/hg19. Variant filtering, annotation, and interpretation were performed using validated bioinformatic pipelines and public clinical and population databases, including ClinVar and gnomAD. Splicing impact was assessed using SpliceAI and Pangolin. Variant-level classification was performed according to ACMG/AMP criteria, while separating variant pathogenicity from disease causality in this specific family.
Molecular Analysis
Next-generation sequencing identified a heterozygous DSC2 variant, c.354+1G>T (NM_024422.6), located at the +1 position of the canonical donor splice site. The variant is rare or absent in population databases and is listed in ClinVar as likely pathogenic, Variation ID 1182317 (Landrum et al., 2018).
In silico prediction supported an effect on splicing, with SpliceAI predicting donor loss, delta score 0.96 (Jaganathan et al., 2019), and Pangolin predicting splice loss, score 0.64 (Zeng and Li, 2022). The variant also showed a CADD PHRED score of 33 and a PhyloP score of 8.87, consistent with high predicted deleteriousness and conservation of the affected region.
Family testing confirmed the presence of the DSC2 c.354+1G>T variant in the proband, her mother, and her daughter, indicating vertical transmission across three generations. The proband had hypertrophic cardiomyopathy and syncope, while her mother had a cardiac conduction disorder requiring pacemaker implantation. The daughter was identified as a carrier; no definite arrhythmogenic cardiomyopathy phenotype was documented in the available clinical data.
At the variant level, the main ACMG/AMP evidence includes PVS1, due to disruption of a canonical splice donor site in a gene associated with inherited cardiomyopathy, and PM2_supporting, due to absence or extreme rarity in population databases (Richards et al., 2015; Karczewski et al., 2020). External clinical database support is limited because ClinVar reports the variant as likely pathogenic based on a single-submitter entry (Landrum et al., 2018). Segregation evidence remains limited and should be interpreted cautiously because of the small number of informative carriers and the heterogeneous phenotypes observed in the family.
Table 1: Molecular and functional characterization of the DSC2 c.354+1G>T germline variant.
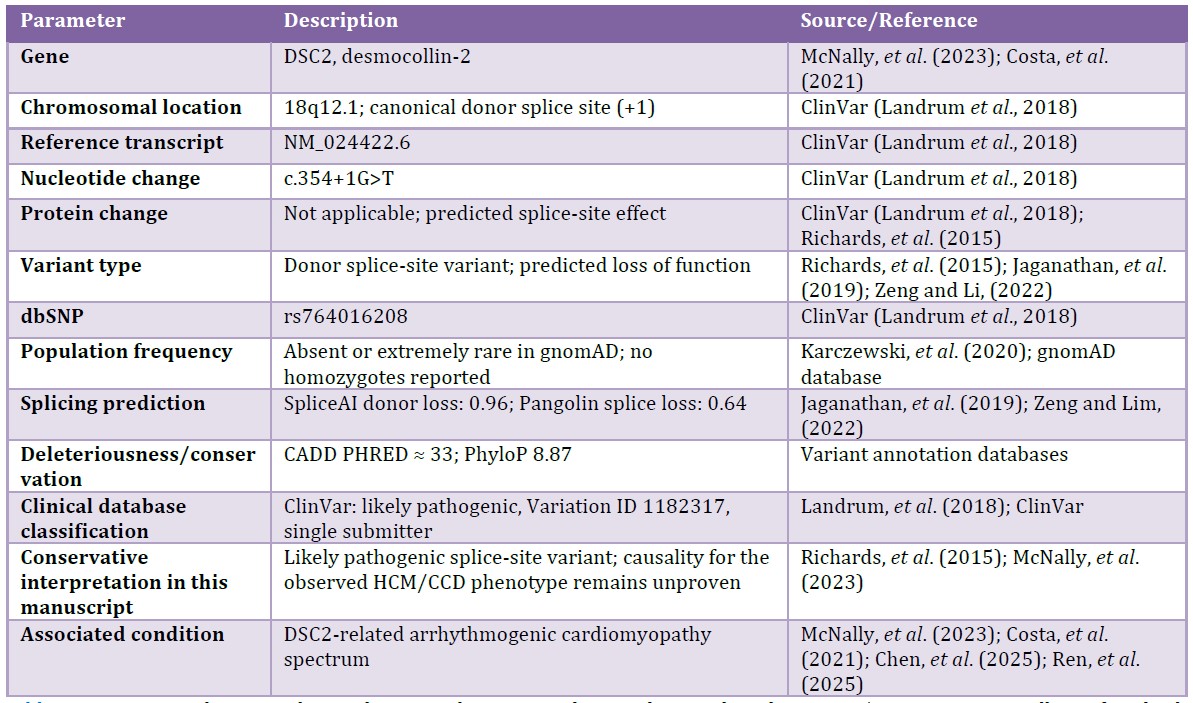
Abbreviations: HCM, hypertrophic cardiomyopathy; CCD, cardiac conduction disorder; ACMG/AMP, American College of Medical Genetics and Genomics/Association for Molecular Pathology.
Discussion
This report describes the familial characterization of the DSC2 c.354+1G>T splice-site variant in a Panamanian family evaluated for suspected inherited cardiac disease. The index case was a 66-year-old woman with a clinical diagnosis of hypertrophic cardiomyopathy, concentric hypertrophy on echocardiography, and high-risk syncope. Her mother, aged 86 years, had cardiac conduction disease requiring pacemaker implantation. At the molecular level, the variant is located at the canonical +1 donor splice site of DSC2, a gene encoding desmocollin-2, a desmosomal cadherin involved in cardiomyocyte adhesion at the intercalated disc. Disruption of desmosomal proteins has been associated with arrhythmogenic cardiomyopathy and related inherited cardiac phenotypes. In this family, the variant showed a SpliceAI donor-loss score of 0.96, a Pangolin splice-loss score of 0.64, a CADD PHRED score of 33, and a PhyloP conservation score of 8.87. ClinVar classifies the variant as likely pathogenic, and ACMG/AMP interpretation supports a likely pathogenic classification mainly based on PVS1 and PM2_supporting.
The available clinical data support cardiovascular surveillance in relatives carrying or at risk of carrying the variant. However, the available records did not document a definitive arrhythmogenic right ventricular cardiomyopathy phenotype in the proband or relatives. Specifically, there were no available data confirming characteristic electrocardiographic abnormalities, documented ventricular arrhythmias, right ventricular structural abnormalities, or cardiac magnetic resonance findings compatible with fibrofatty replacement or myocardial fibrosis in a typical distribution. Therefore, the observed phenotype should be described as heterogeneous inherited cardiac disease rather than confirmed arrhythmogenic cardiomyopathy.
The observation of vertical transmission provides useful information for family counseling and cascade testing. Nevertheless, segregation evidence remains limited because only a small number of informative relatives were available, and the clinical manifestations differed among family members. For this reason, the variant should be interpreted as likely pathogenic at the molecular level, while its role as the sole cause of hypertrophic cardiomyopathy or conduction disease in this family remains unproven.
Recent studies have emphasized the clinical relevance of desmosomal genes in inherited cardiac disease. Chen, et al. (2025) reported that variants in DSG2 and DSC2 are associated with right ventricular or biventricular disease and adverse outcomes in arrhythmogenic cardiomyopathy cohorts. Ren, et al. (2025) further highlighted the role of desmocollin-2 in desmosomal remodeling in arrhythmogenic cardiomyopathy. In addition, Wu, et al. (2022) and Lin, et al. (2021) described rare desmosomal variants in patients with hypertrophic or overlapping cardiomyopathy phenotypes. These findings support the relevance of DSC2 in inherited cardiac disease, but they also indicate that additional genetic, epigenetic, or environmental contributors should be considered when the phenotype is atypical.
The same variant was previously reported in a broader Panamanian cohort of hereditary cardiomyopathies (Cedeño-Escudero et al., 2026). The added value of the present report is the family-oriented characterization of the variant, the conservative interpretation of genotype-phenotype correlation, and the emphasis on expanded phenotyping and longitudinal follow-up rather than a claim of a definitive arrhythmogenic cardiomyopathy family.
From a clinical perspective, the identification of this variant supports genetic counseling, cascade testing, and cardiological surveillance in carriers and at-risk relatives. Recommended phenotyping should include detailed electrocardiography, Holter monitoring, echocardiography, exercise testing when appropriate, and cardiac magnetic resonance imaging to evaluate right ventricular involvement, biventricular disease, myocardial fibrosis, and arrhythmic risk. These data would be essential to strengthen genotype-phenotype correlation in future studies.
Limitations
This study has several limitations. First, the number of genetically informative family members is small, limiting segregation analysis. Second, the available clinical information does not establish a definite diagnosis of arrhythmogenic right ventricular cardiomyopathy in the proband or relatives. Third, detailed electrocardiography, Holter monitoring, cardiac magnetic resonance imaging, and longitudinal follow-up data were not available for all carriers. Fourth, no functional RNA or protein study was performed to confirm the predicted splicing defect. Finally, additional genetic contributors cannot be excluded, particularly given the hypertrophic cardiomyopathy phenotype in the proband and the conduction disorder in the mother.
Conclusions
The DSC2 c.354+1G>T variant is a rare canonical splice-site variant with strong computational evidence for altered splicing and relevance to the desmosomal cardiomyopathy spectrum. In this Panamanian family, the variant was observed across three generations, supporting cascade testing and clinical surveillance. However, the currently available data do not prove that this variant is the sole cause of the hypertrophic cardiomyopathy and conduction disease observed in the family. Expanded phenotyping and family testing are required to clarify its clinical significance.
Ethics Approval and Consent for Publication: Ethical approval for the original genetic study was granted by the Institutional Research Ethics Committee of the Social Security Fund, Panama, under protocol number DENSYPS-DENADOI-N-404-2024. Access to anonymized genomic data was authorized by the National Specialized Center for Medical Genetics and Genomics, Ciudad de la Salud, Panama. The present study did not involve additional patient recruitment or biological sample collection.
Written informed consent for publication of the clinical, genetic, and pedigree information was obtained from the patient or legal representative, as applicable. The signed informed consent form is submitted together with the revised manuscript. All data were handled according to ethical and confidentiality considerations.
Conflict of Interest: All authors declare that they participated in the design, execution, and analysis of this study and approved the final version of the manuscript. The authors declare no conflicts of interest related to this article.
Data Availability: The data supporting the findings of this study are included in this article. Additional information is available from the corresponding author upon reasonable request, subject to ethical and confidentiality considerations.
References:
Cedeño-Escudero JA, Sotillo-Bent LA, Medina-Batista E, Méndez-Rosado LA. Genetic Variants in Panamanian Patients with Hereditary Cardiomyopathies. OBM Genetics 2026; 10: 1-3.
Chen L, Hu Y, Saguner AM, Bauce B, Liu Y, Shi A, Guan F, Chen Z, Bueno Marinas M, Wu L, Foltran D, Hermida A, Fressart V, Pinci S, Celeghin R, Chen Z, Zhang B, Lin Y, Liu X, Cason M, Martini M, Rigato I, Brunckhorst C, Biller R, Basso C, Yang B, Zhao X, Cadrin-Tourigny J, Gasperetti A, James CA, Zhou X, Gandjbakhch E, Pilichou K, Duru F, Hu S. Natural History and Clinical Outcomes of Patients With DSG2/DSC2 Variant-Related Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2025; 151: 1213-1230.
Costa S, Cerrone M, Saguner AM, Brunckhorst C, Delmar M, Duru F. Arrhythmogenic cardiomyopathy: An in-depth look at molecular mechanisms and clinical correlates. Trends Cardiovasc Med 2021; 31: 395-402.
Goudal A, Karakachoff M, Lindenbaum P, Baron E, Bonnaud S, Kyndt F, Arnaud M, Minois D, Bourcereau E, Thollet A, Deleuze JF, Genin E, Wiart F, Pasquié JL, Galand V, Sacher F, Dina C, Redon R, Bezieau S, Schott JJ, Probst V, Barc J. Burden of rare variants in arrhythmogenic cardiomyopathy with right dominant form-associated genes provides new insights for molecular diagnosis and clinical management. Hum Mutat 2022; 43: 1333-1342.
Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, Chow ED, Kanterakis E, Gao H, Kia A, Batzoglou S, Sanders SJ, Farh KK. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019; 176: 535-548.e24.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O'Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME; Genome Aggregation Database Consortium; Neale BM, Daly MJ, MacArthur DG. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020 May;581(7809):434-443. doi: 10.1038/s41586-020-2308-7. Epub 2020 May 27. Erratum in: Nature. 2021 Feb;590(7846):E53. doi: 10.1038/s41586-020-03174-8. Erratum in: Nature 2021; 597: E3-E4.
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, Karapetyan K, Katz K, Liu C, Maddipatla Z, Malheiro A, McDaniel K, Ovetsky M, Riley G, Zhou G, Holmes JB, Kattman BL, Maglott DR. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018; 46: D1062-D1067.
Lin Y, Huang J, Zhu Z, Zhang Z, Xian J, Yang Z, Qin T, Chen L, Huang J, Huang Y, Wu Q, Hu Z, Lin X, Xu G. Overlap phenotypes of the left ventricular noncompaction and hypertrophic cardiomyopathy with complex arrhythmias and heart failure induced by the novel truncated DSC2 mutation. Orphanet J Rare Dis 2021; 16: 496.
McNally E, MacLeod H, Dellefave-Castillo L. Arrhythmogenic Right Ventricular Cardiomyopathy Overview. 2005 Apr 18 [updated 2023 May 11]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2026. PMID: 20301310.
Ren J, Hu Z, Wu L, Akdis D, Ye W, Saguner AM, Su M, Dong H, Chen Z, Hu D, Hu S, Duru F, Chen L. Comprehensive analysis of desmosomal protein remodelling identifies desmocollin-2 as potential biomarker for arrhythmogenic cardiomyopathy. Europace 2025; 27: euaf199.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405-424.
Wu G, Liu J, Ruan J, Yu S, Wang L, Zhao S, Wang S, Kang L, Wang J, Song L. Deleterious Rare Desmosomal Variants Contribute to Hypertrophic Cardiomyopathy and Are Associated with Distinctive Clinical Features. Can J Cardiol 2022; 38: 41-48.
Zeng T, Li YI. Predicting RNA splicing from DNA sequence using Pangolin. Genome Biol 2022; 23: 103.
|
